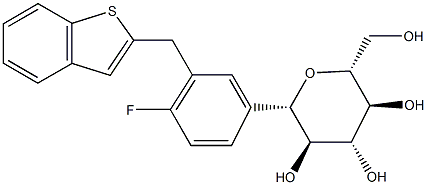
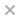体外研究
纳摩尔浓度的Ipragliflozin可浓度依赖性地抑制小鼠、大鼠和人源SGLT2的活性。此外,ipragliflozin不抑制人源SGLT4和SGLT5(IC50>1,000 nM)。在小鼠3T3-L1、大鼠L6、人源Caco-2和HepG2细胞中,ipragliflozin也不抑制某些GLUT亚型,如GLUT1和GLUT4(IC50>1,000 nM)。Ipragliflozin不与各种受体、离子通道、转运体相互作用,如肾上腺素受体(α1, α2, β), 毒蕈碱型受体(如M1, M2), 血管紧缩素(AT1和AT2), 钙离子通道(L-type和N-type), 钾离子通道(KATP和SKCa), 钠离子通道(site 2), 肠促胰酶肽(CCKA和CCKB), 多巴胺(如D1, D2), 内皮素(ETA和ETB), γ-氨基丁酸(GABAA和GABAB), 谷氨酸盐(AMPA, kainate和NMDA), 血清素(5-HT1, 5HT2B, and transporter), 组胺(H1, H2, and H3)和神经激肽(NK1, NK2, and NK3),IC50>3000 nM。Ipragliflozin不受小鼠肠道的葡糖苷酶影响,保持稳态。